
Progeria (Syndrom Hutchinsona-Gilforda) „przedwczesna starość” zaliczana jest do bardzo rzadko występujących chorób genetycznych. Choroba ta polega na szybko postępującym, niemożliwym do zatrzymania procesie starzenia się organizmu. W chwili obecnej nie istnieją sposoby na zatrzymanie i leczenie tego procesu a sama przyczyna choroby nie została dokładnie wyjaśniona.
Progeria (Syndrom Hutchinsona-Gilforda)
Co to jest progeria?
Nazwa syndromu progerii Hutchinsona-Gilforda pochodzi od nazwisk dwóch brytyjskich lekarzy, doktora Jonathana Hutchinsona i Hastingsa Gilforda, którzy prowadzili nad nim badania, stąd też medyczne określenie schorzenia – zespół progerii Hutchinsona-Gilforda ( Hutchinson-Gilford Progeria Syndrome – HGPS). Progeria to choroba genetyczna. Jej istotą jest niezwykle szybki proces starzenia się organizmu.
Przyczyna choroby do tej pory nie została wyjaśniona. Progeria po raz pierwszy została opisana przez Jonathana Hutchinsona w 1886 roku,a następnie przez chirurga Hastingsa Gilforda. Jest to choroba występująca bardzo rzadko. Syndrom ten występuje u małych dzieci. Pierwsze objawy widoczne są, gdy dziecko skończy pierwszy rok swojego życia. Pełny objaw choroby widoczny jest po około ukończeniu 2 roku życia przez dziecko. Zespół dotyka dzieci obojga płci, a co ważne, wśród rodzeństwa jedno może być chore, a drugie zdrowe.
Za proces powstawania progerii odpowiedzialne są mutacje białek zawartych w jądrach komórkowych, tzw. lamin. Białka te odpowiadają w jądrach komórkowych m.in. za regulację procesu mitozy. Mutacja ta prowadzi do produkcji zmutowanej proteiny (tzw. progerin), która jest odpowiedzialna za zniekształcenie jąder komórkowych. Konsekwencją tego procesu jest zaprzestanie produkcji substancji międzykomórkowej w komórkach tkanki łącznej. Objawia się to silnymi zaburzeniami budowy i działania układu szkieletowego, skóry i układu naczyniowego. Brak produkcji substancji międzykomórkowej powoduje, że komórki przestają się dzielić i wskutek tego umierają.
Progeria - rozwój choroby
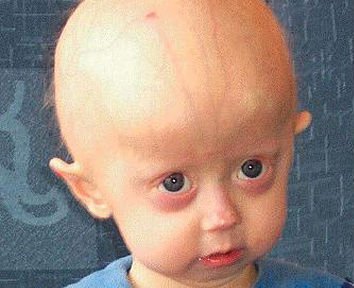
W momencie urodzenia i w czasie niemowlęctwa dzieci wyglądają normalnie, mają zazwyczaj prawidłową masę ciała, a choroba przebiega bezobjawowo. W niektórych jednak przypadkach można zauważyć pewne charakterystyczne cechy twarzy (sinica w środkowej części i tzw. rzeźbiony nos). Czasem widoczna jest nieustająca wysypka i zaczerwienienia na skórze.
W drugim roku życia wygląd ulega drastycznym zmianom. Skóra malucha wiotczeje, tworzą się na niej zmarszczki, jest sucha, wypadają włosy, brwi i rzęsy, dziecko ząbkuje z opóźnieniem, nie rośnie i nie przybiera prawidłowo na wadze. Dodatkowo zauważalne jest to, iż głowa jest bardzo dużych rozmiarów w porównaniu do reszty ciała.
Dziecko jest niskie, bardzo szczupłe, ma wyłupiaste oczy, klatka piersiowa ma nieregularny kształt, obojczyki są krótkie, biodra koślawe, skóra jest cienka, sucha i przeźroczysta. Kości kończyn stają się powykrzywiane, bardzo podatne na złamania. Z czasem maluch ma problemy z poruszaniem się, wyprostowaniem kończyn górnych i dolnych. Nie dojrzewa płciowo. Wszystkie procesy patologiczne zachodzą w organizmie dziecka bardzo szybko i są identyczne z tymi, które dotykają osoby w starszym wieku.
Rozwój umysłowy przebiega bez zakłóceń, a czasami obserwuje się nawet ponadprzeciętne uzdolnienia. Maluch nie ma problemów ze wzrokiem, słuchem oraz charakterystycznych dla osób starszych zmian osobowości.
Osoby dotknięte progerią żyją średnio 13 lat. Wyglądem przypominają oni „małych starców”.
U chorych na progerię w wieku dwóch lat:
- na twarzy pojawiają się zmarszczki,
- skóra staje się sucha i pomarszczona,
- postępuje utrata włosów, rzęs oraz brwi,
- oczy stają się podkrążone, wyłupiaste,
- pod oczami pojawiają się fałdy skóry.
Główka dziecka jest bardzo duża w porównaniu do reszty ciała i widoczne są na niej silnie zarysowane żyły. Policzki na twarzy zapadają się, stawy sztywnieją. „Mali starcy” mają problem z wyprostowaniem nóg bądź rąk. Kości stają się bardzo podatne na złamania, jak w przypadku osób starszych. Z czasem dochodzi do rozwoju chorób charakterystycznych dla osób wieku starszego, takich jak osteoporoza, choroby układu krążenia, problemy z układem pokarmowym czy oddechowym. Ciało osoby chorej ulega zmianie nawet o 10 lat na każdy rok życia.
Chorzy umierają najczęściej z powodu udaru mózgu bądź zawału serca – charakterystyczne są zmiany miażdżycowe w naczyniach krwionośnych. Występuje u nich także zwiększone ryzyko wystąpienia nowotworów.
Objawy progerii
Wśród objawów progerii wyróżnia się:
- łysienie,
- sztywność skóry,
- cienka skóra z brązowymi plamami z zmianami oraz zmarszczkami, silnie przesuszona,
- niska masa ciała,
- napięcie mięśniowe,
- głowa nieproporcjonalnie duża do reszty ciała,
- wyłupiaste oczy,
- późne wyrastanie zębów lub brak ich wyrastania,
- koślawe biodra,
- chód na szerokiej podstawie,
- wychudzone kończyny, podatne na złamania,
- „ptasi” koniec nosa,
- utrata tkanki podskórnej,
- utrata brwi oraz rzęs – uogólnione łysienie,
- niski wzrost,
- niedorozwój żuchwy,
- gruszkowata klatka piersiowa,
- krótkie obojczyki,
- dystroficzne paznokcie,
- pojawienie się owrzodzeń,
- miażdżyca,
- brak rozwoju płciowego.
Bardzo ważną rolę odgrywa przeprowadzenie szczegółowych badań laboratoryjnych, zwłaszcza moczu i krwi. Wykazują one najczęściej silnie podwyższony poziom trójglicerydów we krwi oraz kwasu hialuronowego w moczu. Szczegółowo oceniana jest praca serca (elektrokardiografia i echokardiografia) oraz przeprowadzane jest badanie radiologiczne kości.
Leczenie progerii
W chwili obecnej nie ma skutecznego i pewnego sposobu leczenia progerii. Szczególną rolę odgrywa regularna kontrola ogólnego stanu zdrowia pacjenta oraz wykonywanie niezbędnych badań. Leczenie ma jedynie charakter objawowy i wymaga współpracy wielu lekarzy specjalistów.
Pacjenci powinni być pod stałą opieką lekarską ze względu na możliwość wystąpienia problemów z układem krążenia, pojawienia się nowotworów, trudno gojących się owrzodzeń. Niestety, nie znaleziono jeszcze sposobu leczenia progerii. Postępowanie terapeutyczne ma jedynie charakter objawowy.
Czytaj również: Nerwica depresyjna – przyczyny, objawy, leczenie nerwicy depresyjnej
Bibliografia
W Wylecz.to opieramy się na EBM (Evidence Based Medicine) – medycynie opartej na faktach i wiarygodnych źródłach. Dowiedz się więcej o tym, jak dbamy o jakość naszych treści.
- Atlas neuroanatomii i neurofizjologii Nettera, David L. Felten, Anil N. Shetty. Wyd. 2012 r.
- Diagnostyka obrazowa. Układ nerwowy ośrodkowy, Joanna Bladowska, Agnieszka Pomianowska, Katarzyna Sklinda, Jerzy Walecki. Wyd. 2014 r.
- Anatomia narządów wewnętrznych i układu nerwowego człowieka, Zofia Ignasiak. Wyd. 2014 r.
Opublikowano: 04.11.2013; aktualizacja: 12.02.2018
Oceń:
4.4
Ten tekst został napisany przez Eksperta, który posiada kierunkowe wykształcenie oraz doświadczenie. Nawiązując współpracę z autorytetami w danej dziedzinie nauki serwis Wylecz.to dba o najwyższą jakość i wiarygodność informacji publikowanych w serwisie. W Wylecz.to opieramy się na EBM (Evidence Based Medicine) – medycynie opartej na faktach i wiarygodnych źródłach.

Magdalena Kacperska
Lekarz
Dr n. med. Magdalena Justyna Kacperska, studia magisterskie ukończyła w 2009 roku na Wydziale Biologii i Ochrony Środowiska. Stopień naukowy dr n.med. obroniła z wyróżnieniem na Wydziale Wojskowo-Lekarskim Uniwersytetu Medycznego w Łodzi w 2014 roku. Posiada liczne publikacje w języku polskim oraz języku angielskim. Zaangażowana w liczne projekty naukowe (z zakresu neurologii, immunologii, chirurgii naczyniowej) oraz pracę dydaktyczną. Szczególnie zaangażowana w projekty związane ze stwardnienie rozsianym (SM), rolą miRNA w SM, udarami niedokrwiennymi oraz destabilizacją blaszki miażdżycowej.














Komentarze i opinie (0)