
Zespół Wiskotta-Aldricha to rzadka choroba o podłożu genetycznym, którą częściej diagnozuje się u mężczyzn. Schorzenie dziedziczone jest w sposób recesywny poprzez chromosom X. Objawy choroby Wiskotta-Aldricha to: zmiany skórne, w tym świąd skóry, nawracające infekcje dróg oddechowych oraz niski poziom płytek krwi w badaniu (trombocytopenia). Jak można leczyć opisywany zespół i czy rokowania są pomyślne dla pacjenta?
Zespół Wiskotta-Aldricha – dziedziczenie, objawy, badania, leczenie, rokowanie

Spis treści
- Zespół Wiskotta-Aldricha – dziedziczenie – u kogo występuje?
- Zespół Wiskotta-Aldricha – białko WASp
- Zespół Wiskotta-Aldricha – objawy choroby
- Trombocytopenia w zespole Wiskotta-Aldricha
- Zespół Wiskotta-Aldricha a swędzenie skóry
- Zespół Wiskotta-Aldricha – nawracające infekcje
- Zespół Wiskotta-Aldricha – poziom białych krwinek i przeciwciał
- Zespół Wiskotta-Aldricha – diagnostyka, badania
- Zespół Wiskotta-Aldricha – leczenie
- Zespół Wiskotta-Aldricha – rokowanie
Zespół Wiskotta-Aldricha – dziedziczenie – u kogo występuje?
Wiskott-Aldrich syndrome to niezwykle rzadka choroba posiadająca podłoże genetyczne. Zespół Wiskotta-Aldricha dziedziczony jest w sposób recesywny poprzez chromosom X (tzw. dziedziczenie sprzężone z chromosomem X).
Choroba Wiskotta-Aldricha jest więc dziedziczona podobnie jak hemofilia (niedobór jednego z czynników krzepnięcia) oraz daltonizm (niemożność rozróżniania barw, najczęściej czerwonej i zielonej).
W związku z powyższym, osobami najczęściej chorującymi na zespół Wiskotta są mężczyźni (wystarczy, że przekazana im zostanie przez matkę jedna wadliwa kopia genu zlokalizowana na chromosomie X). Nie oznacza to jednak, że dziewczynki w ogóle nie chorują, jednak jest to zdecydowanie rzadsza sytuacja ze względu na konieczność przekazania dziewczynce wadliwego chromosomu zarówno przez matkę, jak i ojca. Sytuacja taka może mieć miejsce, gdy matka dziewczynki jest tzw. nosicielką – posiada jedną wadliwą kopię genu oraz jedną zdrową kopię genu (jeden z jej chromosomów jest zdrowy, a drugi ma wyżej wymieniony defekt) lub choruje, a ojciec dziecka również boryka się ze schorzeniem. Przypadki takie są jednak bardzo rzadkie i zazwyczaj nie spotyka się ich w codziennej praktyce lekarskiej.
Zgodnie z danymi pochodzącymi od różnych autorów, zespół Wiskotta-Aldricha występuje z częstością 3–4 przypadków na 1 milion żywych urodzeń. Schorzenie zalicza się do grupy chorób rzadkich.
Zespół Wiskotta-Aldricha – białko WASp
Zespół Wiskotta to, jak już zostało wcześniej wspomniane, choroba genetyczna dziedziczona z chromosomem X. Wadliwa budowa genu WAS (zlokalizowanego na chromosomie X) sprawia, że białko WASp nie jest w życiu pozałonowym produkowane lub jego produkcja nie jest wystarczająca. Białko to (nazywane również białkiem Aldricha i Wiskotta) niezbędne jest do przeprowadzania prawidłowych procesów reorganizacji cytoszkieletu komórek ludzkiej krwi.
Ponadto WAS protein bierze udział w programowanej śmierci komórki (tj. apoptozie) i przekazywaniu sygnałów pomiędzy komórkami. Białko to związane jest z komórkami takimi jak limfocyty oraz płytki krwi.
Zespół Wiskotta-Aldricha – objawy choroby
Uwarunkowany genetycznie zespół Wiskotta-Aldricha może ujawnić się zarówno w życiu niemowlęcym, jak i bezpośrednio po urodzeniu. Należy podkreślić, że stopień ciężkości objawów zależny jest od rodzaju mutacji, która obserwowana jest w obrębie genu kodującego białko WAS.
W przypadku pojedynczych, punktowych mutacji możemy mówić nie o zespole Wiskotta-Aldricha, lecz o trombocytopenii sprzężonej z chromosomem X. Wówczas nasilenie objawów, takich jak obniżenie odporności czy tez wyprysk skórny, jest mniejsze. W tej odmianie choroby obserwuje się także niższe ryzyko wystąpienia nowotworów.
Obniżona produkcja białka WASp lub jego nieprawidłowa budowa jest każdorazowo związana z wystąpieniem pełnoobjawowego zespołu Wiskotta-Aldricha. Obserwowana jest wówczas bardzo charakterystyczna triada objawów, do której zaliczamy:
- trombocytopenię (obniżona liczba płytek krwi, czyli trombocytów),
- wyprysk skórny,
- nawracające infekcje (szczególnie dróg oddechowych).
Ponadto wystąpić mogą inne, mniej charakterystyczne objawy. Na szczególną uwagę zasługują tutaj choroby o podłożu autoimmunologicznym (tzw. choroby z autoagresji), czyli niedokrwistość hemolityczna o podłożu immunologicznym oraz choroby nerek.
Trombocytopenia w zespole Wiskotta-Aldricha
Trombocytopenia, czyli zmniejszona ilość płytek krwi to jeden z głównych objawów zespołu Wiskotta-Aldricha. W początkowym stadium choroby manifestuje się ona m.in. niewielkimi wybroczynami na skórze i zwiększoną skłonnością do siniaków. Ponadto występują krwiste biegunki (często jest to pierwszy objaw choroby, występujący jeszcze w okresie noworodkowym).
W późniejszym życiu chory boryka się m.in. z krwawieniami dziąseł, wylewami podskórnymi oraz nawracającymi krwawieniami z nosa. Rzadziej występują krwawienia wewnętrzne (np. do jamy brzusznej).
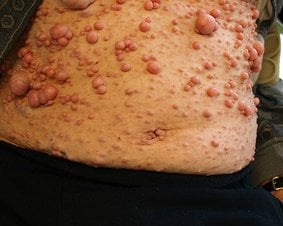
Zespół Wiskotta-Aldricha a swędzenie skóry
W zespole Wiskotta-Aldricha zmiany skórne są bardzo zróżnicowane nie tylko ze względu na lokalizację, ale również ich wygląd. Pierwsze manifestacje skórne zazwyczaj pojawiają się w wieku niemowlęcym. Wówczas mogą one przypominać zmiany odparzeniowe oraz oparzenia. Często są one bagatelizowane ze względu na swój łagodny przebieg (bardzo mylący jest tutaj brak współwystępowania innych objawów charakterystycznych dla zespołu Wiskotta-Aldricha). Stosowanie leków uśmierzających świąd nie zawsze jest skuteczne.
Jedną z najbardziej charakterystycznych cech zmian skórnych jest ich swędzenie. W wyniku drapania, w początkowej fazie powstają przeczosy, a następnie trudno gojące się rany. Ponadto obserwuje się zwiększoną skłonność do zakażeń skóry. Infekcje te są zazwyczaj trudne w leczeniu i wymagają często doustnej antybiotykoterapii.
Zespół Wiskotta-Aldricha – nawracające infekcje
Jednym z najbardziej charakterystycznych objawów zespołu Wiskotta-Aldricha są nawracające infekcje. Brak lub niedobór (a także nieprawidłowa budowa) białka WAS powoduje obniżenie liczby limfocytów zarówno grupy T, jak i B. W mniej nasilonych odmianach choroby liczba limfocytów B może być prawidłowa, jednak wówczas zazwyczaj dochodzi do upośledzenia ich funkcjonalności.
Obniżona liczba limfocytów T szczególnie predysponuje chorych do zapadania na infekcje wirusowe. Zakażenia wirusowe najczęściej obserwowane w zespole Wiskotta-Aldricha to: grypa, wirusowe zapalenie płuc i oskrzeli, a także nawracające zapalenia ucha środkowego. Ponadto należy wspomnieć o nawracającym zakażeniu wirusem opryszczki ludzkiej – tzw. zimno pojawia się bez względu na porę roku.
W związku z obniżoną liczbą lub nieprawidłową funkcją limfocytów B u chorych bardzo często stwierdzamy zwiększoną zachorowalność na zakażenia bakteryjne. Osoby te są szczególnie narażone na bakteryjne zapalenia płuc, zapalenie opon mózgowo-rdzeniowych, a także gruźlicę. W literaturze opisywane są również przypadki rzekomobłoniastego zapalenia jelit.
Zespół Wiskotta-Aldricha – poziom białych krwinek i przeciwciał
W podstawowych i specjalistycznych badaniach laboratoryjnych obserwowane są zjawiska takie jak: limfocytopenia (obniżona liczba białych krwinek, czyli limfocytów), nawracające zakażenia, trombocytopenia (zmniejszona liczba płytek krwi, czyli trombocytów) – jej wykładnikiem są krwawienia i wybroczyny.
Ilość przeciwciał poszczególnych klas kształtuje się następująco:
- przeciwciała klasy IgG – ich liczba jest zazwyczaj prawidłowa, ale mogą być również obniżone;
- przeciwciała klasy IgM – ich poziom jest obniżony, są to tzw. przeciwciała I linii (odpowiadające za odporność w początkowym stadium zakażenia);
- przeciwciała klasy IgA – stężenie prawidłowe lub nieznacznie podniesione;
- przeciwciała klasy IgE – stężenie prawidłowe lub nieznacznie podniesione, zbyt duża liczba przeciwciał może wiązać się z reakcjami nadwrażliwości.
Zespół Wiskotta-Aldricha – diagnostyka, badania
Diagnostyka zespołu Wiskotta-Aldricha rozpoczyna się zazwyczaj bezpośrednio po urodzeniu dziecka (najczęściej chłopca). Występowanie w najbliższej rodzinie (w linii męskiej) potwierdzonego zespołu obliguje lekarzy zajmujących się dzieckiem po porodzie do rozpoczęcia postępowania diagnostyczno-terapeutycznego.
Należy podkreślić, że aktualnie jedyną pewną metodą umożliwiającą postawienie diagnozy są badania genetyczne. W postawieniu rozpoznania ważne są również wyżej wymienione objawy, które mają za zadanie nakierować lekarza oraz umożliwić mu przeprowadzenie diagnostyki różnicowej.
Ważną kwestią są badania dodatkowe. Zaliczamy do nich m.in. badania laboratoryjne krwi oraz biopsję szpiku. Biopsja szpiku wykonywana jest bardzo często bezpośrednio po postawieniu diagnozy. Jej celem jest określenie stopnia nasilenia procesów hematopoezy, które mają miejsce w szpiku kostnym. Otrzymane dane są również pomocne w jednym z etapów leczenia, tj. podczas przeszczepienia szpiku kostnego.
Zespół Wiskotta-Aldricha – leczenie
Leczenie zespołu Wiskotta-Aldricha w znacznym stopniu poprawia rokowanie chorych, jednak bardzo rzadko prowadzi ono do ich całkowitego wyleczenia. Aktualnie jedną z najczęściej stosowanych metod jest przeszczepienie szpiku kostnego, czyli przeszczep komórek hematopoetycznych. Metoda ta, pomimo najlepszych rokowań, nie zawsze jest możliwa do wykonania ze względu na niemożność dobrania odpowiedniego dawcy komórek.
Nadzieją dla osób chorujących na zespół Wiskotta-Aldricha jest jednak nie tylko przeszczep rodzinny, ale również skorzystanie z bazy dawców szpiku. Przed przeszczepem szpiku wymagana jest chemioterapia lub radioterapia, ze względu na konieczność zniszczenia wszystkich limfocytów osoby chorej.
W przypadku niemożności wykonania przeszczepu komórek macierzystych stosuje się tzw. leczenie zachowawcze. Działanie to obejmuje:
- regularne przetaczanie koncentratów płytek krwi (w celu wyrównania trombocytopenii),
- dożylną podaż immunoglobulin (czyli wcześniej wymienionych przeciwciał),
- antybiotykoterapię (w przypadku wystąpienia infekcji bakteryjnych),
- leczenie glikokortykosteroidami – zarówno w postaci maści, jak i ogólnie.
Terapia zespołu Wiskotta-Aldricha może obejmować również usunięcie śledziony, która bardzo często w przebiegu tego zespołu bierze udział w niszczeniu płytek krwi. Splenektomia nie pozostaje jednak bez wpływu na organizm człowieka. Usunięcie śledziony może być przyczyną większej ilości nawracających zakażeń (szczególnie bakteryjnych). Podczas prowadzenia procesu leczniczego zespołu nie należy również zapominać o zwiększonym ryzyku wystąpienia u chorych nowotworów i chorób autoimmunologicznych. Zaleca się, by każda osoba chora była poddawana regularnym badaniom, które mają na celu wykluczenie nowotworów (szczególnie białaczek).
Podczas leczenia zespołu Wiskotta-Aldricha nie należy też zapominać o zakazie podawania szczepionek zawierających żywe drobnoustroje. W związku z niemożnością wytworzenia przez organizm przeciwciał, działanie to jest niebezpieczne i może nawet doprowadzić do zgonu osoby zaszczepionej.
Zespół Wiskotta-Aldricha – rokowanie
Przed wprowadzeniem do rutynowej praktyki przeszczepu komórek hematopoetycznych, średni wiek życia pacjentów z zespołem Wiskotta-Aldricha nie przekraczał 3–4 lat. Prawidłowo przeprowadzony przeszczep, a także stosowanie leczenia wspomagającego może wydłużyć czas przeżycia nawet pięciokrotnie.
Nadziei na dłuższe życie osób chorych na zespół Wiskotta-Aldricha upatruje się w terapii genowej. Aktualnie prowadzone badania dają bardzo obiecujące wyniki.
Bibliografia
W Wylecz.to opieramy się na EBM (Evidence Based Medicine) – medycynie opartej na faktach i wiarygodnych źródłach. Dowiedz się więcej o tym, jak dbamy o jakość naszych treści.
- https://primaryimmune.org/sites/default/files/publications/IDF-Patient-Family-Handbook-5th-Edition-2015-Reprint.pdf
- Schwinger W. i wsp., The phenotype and treatment of WIP deficiency: literature synopsis and review of a patient with pre-transplant serial donor lymphocyte infusions to eliminate CMV, Front. Immunol., 2018, 9: 2554.
- Sereni L., Platelets in Wiskott-Aldrich syndrome: victims or executioners? J. Leukoc. Biol., 2018, 103, 3: 577–590.
- Buchbiner D., Wiskott–Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl. Clin. Genet., 2014, 7: 55–66.
Opublikowano: 07.12.2018; aktualizacja: 07.12.2018
Oceń:
4.6














Komentarze i opinie (1)
opublikowany 20.02.2024